癌细胞和免疫细胞围绕营养物质展开的竞争,在肿瘤微环境里可以说无处不在,谁“多吃一口”谁就可能占到上风,而大多数时候受伤的都是免疫细胞。受伤归受伤,T细胞们顶多就是战斗力下降,但最新研究就见到了直接“叛变”帮助癌细胞的,简直是太没骨气了。
上海交通大学李晓光、王慧团队与国内研究者合作在Gut期刊发表的一项最新研究成果显示:浸润到肝癌微环境中的髓系细胞在竞争谷氨酰胺中败给癌细胞后,自身内质网稳态就会被打破,使IRE1α/XBP1信号诱导G蛋白偶联受体GPR109A表达上调,髓系细胞由此变成了免疫抑制功能极强的癌症帮凶,因此GPR109A是具有干预价值的免疫代谢检查点[1]。
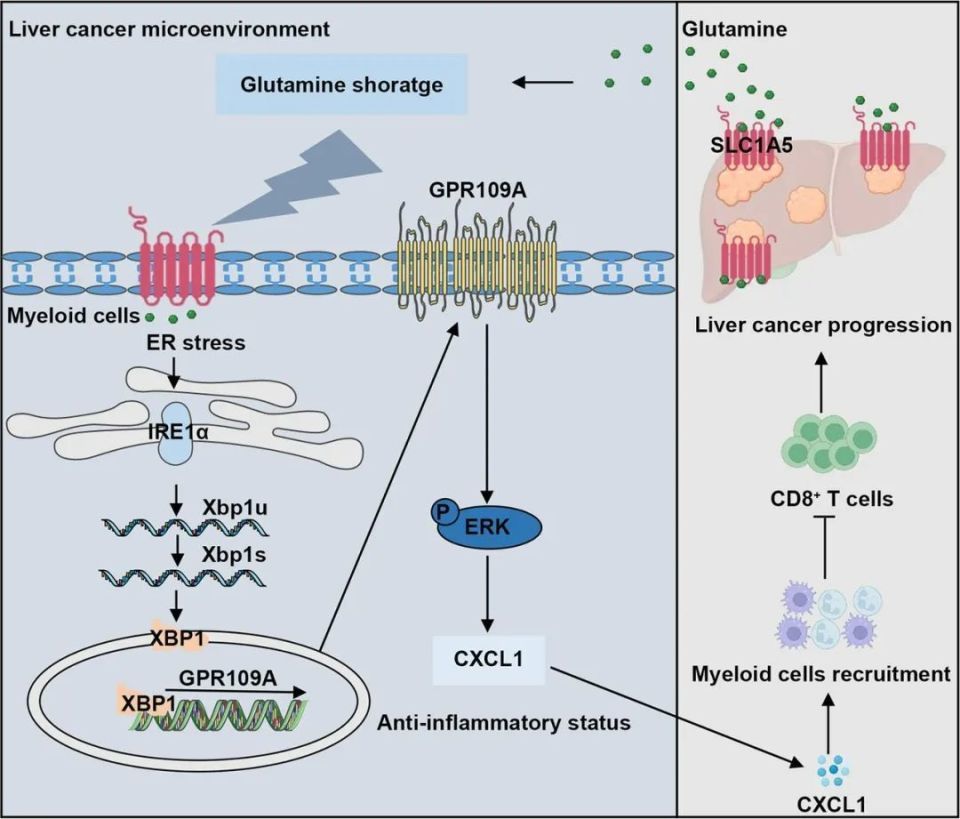
论文核心内容总结
肝癌的免疫微环境是出了名的抑制性极强,也使免疫治疗在此前很长一段时间没能取得突破,必须靠靶免联合方案(PD-1/L1抑制剂+VEGF通路靶向药)破局,而髓系细胞往往就是执行免疫抑制的打手,不管是以髓系来源的免疫抑制细胞(MDSCs)还是以M2型肿瘤相关巨噬细胞(TAMs)形态存在,它们都是免疫细胞和免疫治疗的敌人[2]。
上海交通大学团队在本次研究中,首先借助外部数据库资料和本地肝癌患者队列,证实乙肝病毒相关肝细胞癌(HBV-HCC)大多存在GPR109A的表达上调,且GPR109A表达上调与患者不良预后相关,但与HBV感染导致的肝炎或其它类型肝炎无关;而在HCC微环境中,表达GPR109A的还真就主要是CD45+CD11b+髓系细胞。
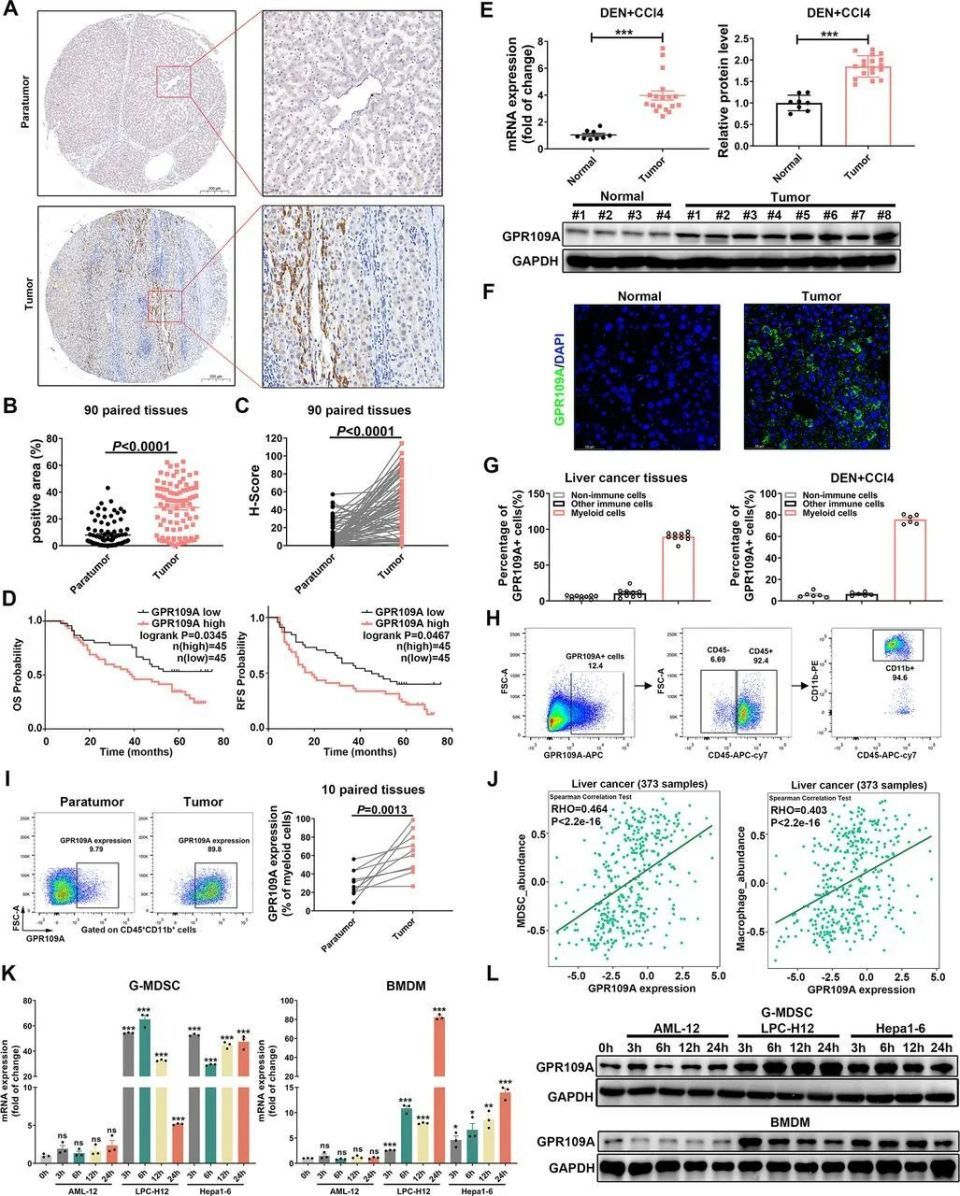
GPR109A表达上调与HCC患者不良预后相关,且GPR109A主要在髓系细胞中表达
研究者们随即用敲除GPR109A的小鼠展开实验,发现肿瘤植入小鼠体内后的生长速度明显偏慢,换句话说就是敲除GPR109A有一定的抑癌效应,而肿瘤微环境中不仅有CD8+T细胞的显著增多,髓系细胞也呈现了正向改变,即MDSCs和M2型TAMs减少而M1型TAMs增多,其它免疫细胞则在敲除GPR109A后基本未受影响。
既然表达GPR109A的主要是髓系细胞,研究者们推测敲除GPR109A的抑癌作用也应该与髓系细胞有关,它们的免疫抑制功能减弱,才让CD8+T细胞成功松绑参与抗癌。为证实这种猜想,研究者们首先测定了微环境内的细胞因子和趋化因子水平,证实多种与髓系细胞免疫抑制功能有关的细胞因子,如CXCL-1、VEGF和IL-10水平均在敲除GPR109A后骤降。
进一步分析显示,敲除GPR109A对MDSCs和TAMs的影响是全方位的,例如这些髓系来源细胞在进入肝癌微环境后会向促癌表型极化,但敲除GPR109A能有效抑制极化的发生,同时还减弱了髓系细胞的迁移能力(主要由CXCL1调控),使它们更难向肿瘤部位浸润和促癌。而反过来,髓系细胞受肝癌微环境的影响上调GPR109A表达,促癌能力就更强了。
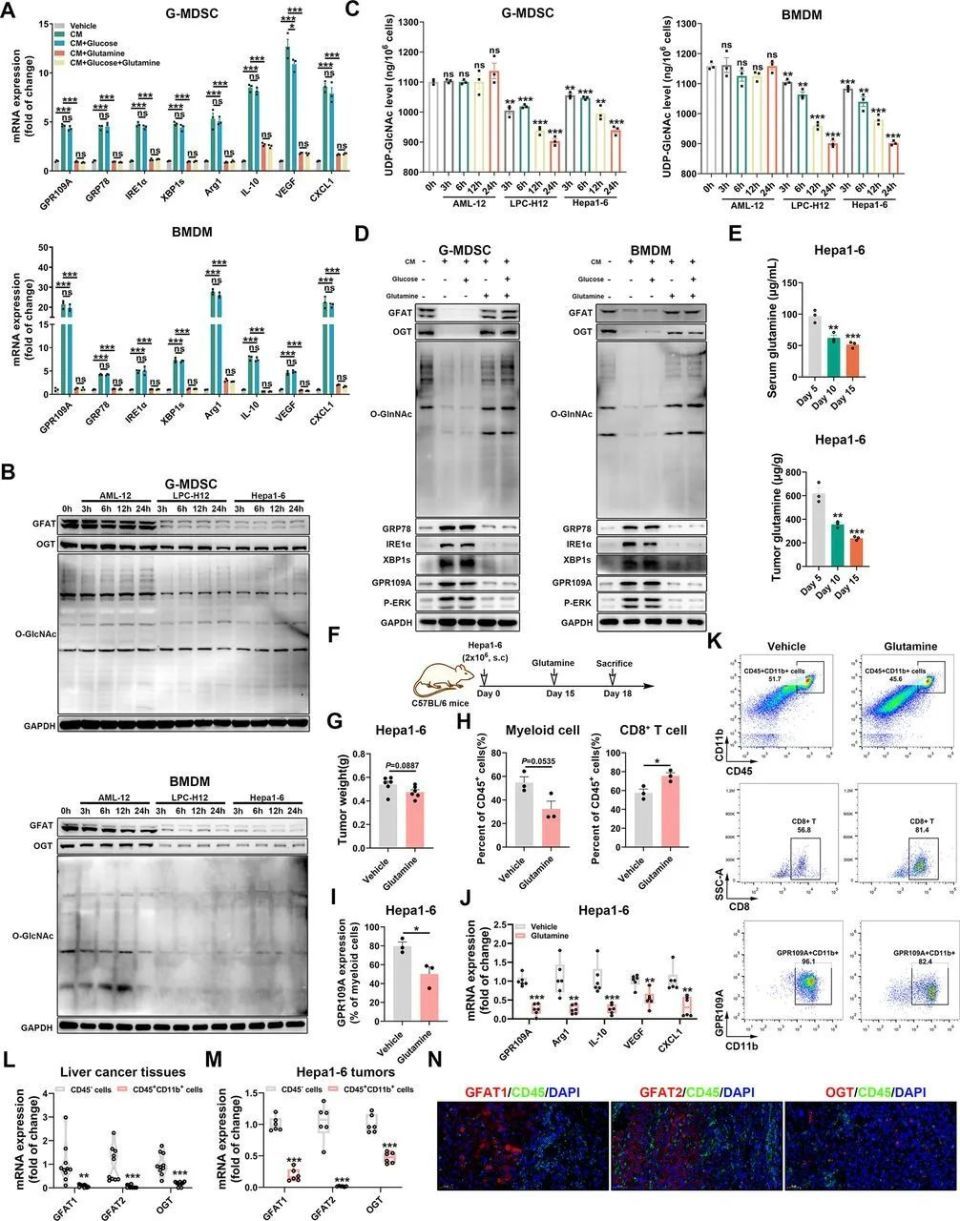
敲除GPR109A可抑制髓系细胞的浸润和向促癌表型极化
那么问题来了,髓系细胞在肝癌微环境中为什么会“堕落”,发生GPR109A的表达上调呢?既往针对寨卡病毒(Zika Virus)感染的一项研究显示,IRE1α/XBP1信号可诱导GPR109A表达上调[3],研究者们也借助单细胞测序确认,作为转录因子的XBP1可结合到编码GPR109A的HCAR2基因启动子部位,以此调控GPR109A表达水平。
正常情况下,IRE1α/XBP1信号对维持细胞内质网稳态有重要作用,它能在这里出现横插一脚,大概率是因为髓系细胞的内质网稳态被肿瘤微环境打破了。借助癌细胞条件培养液(CM)、特定抑制剂和内质网应激诱导剂(thapsigargin),研究者们证实内质网应激确实可以经由磷酸化的ERK激酶激活IRE1α/XBP1信号,进而上调GPR109A表达。
直到这个时候,奇点糕开头提到的谷氨酰胺竞争才终于出场了:实验显示,白介素-2/4等促炎因子处理并不会诱导髓系细胞的内质网应激,但在使用条件培养液处理前先给髓系细胞补充谷氨酰胺,就能预防内质网应激、GPR109A表达上调及随后的表型极化等一系列过程;而HCC微环境中谷氨酰胺的相对短缺,是因为HCC细胞普遍高表达谷氨酰胺转运载体SLC1A5,抢走了更多谷氨酰胺,缺少谷氨酰胺就使髓系细胞沦为了癌症帮凶。
最后,研究者们证实已被识别可阻断GPR109A受体的抗胆碱能药物溴美喷酯(Mepenzolate bromide),能够在细胞和小鼠实验中削弱髓系细胞因GPR109A表达上调而增强的免疫抑制功能,由此激活抗肿瘤免疫应答,并且能够与PD-L1抑制剂治疗协同增效,提供了针对GPR109A这个免疫代谢检查点的潜在免疫联合治疗方案。